GROMACS is a popular open-source program designed explicitly for molecular dynamics simulations that explore the behaviour of complex molecular systems. It is known for its high performance and user-friendly interface, making it an excellent choice for simulating various chemical and biochemical systems, including proteins, nucleic acids, lipids, carbohydrates, and polymers.
GROMACS can be installed and used on various operating systems. The most suitable platform is Linux due to extensive code testing and better performance. However, if you only have a Windows laptop or desktop and only want to practice and learn the program, don’t bother yourself with installing GROMACS on a real Linux OS installed on a physical partition.
You can install and run GROMACS inside your Windows, and if your system doesn’t have a decent CUDA-enabled graphic card, the performance difference between Windows and Linux installation is negligible.
In this tutorial, I will show you how to install GROMACS on Windows 10 and 11 through a feature of widows called Windows Subsystem for Linux (WSL). I will also show you how you can install VMD software and use it alongside the WSL GROMACS. VMD is an excellent molecular visualization program that displays and analyzes molecular systems.
Follow me on LinkedIn
Note: This blog is the text version of the GROMACS installation video tutorial. You can Watch the video here or on the Insilicosci YouTube channel. You can also copy and paste commands from this page if you prefer not to type them.
Installing Ubuntu on Windows Subsystem for Linux
Ubuntu is a widespread Linux distribution. Installing Ubuntu on a Windows system is straightforward. First, enable the Windows subsystem, and then select Ubuntu from the Microsoft store.
Start by pressing the Start button and typing “Control Panel” to open it. Next, click “Programs,” then select “Turn Windows features on or off.” Enable both the Virtual Machine Platform and the Windows Subsystem for Linux in this menu. Finally, click “OK” to apply the changes. After that, restart your computer to make it work.
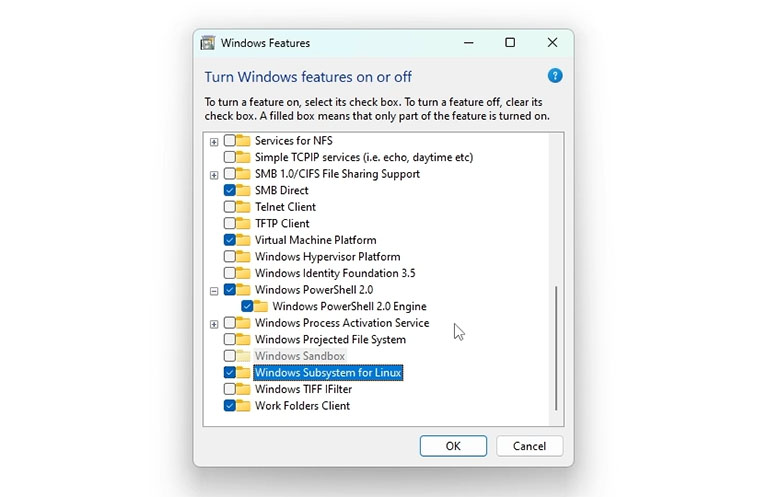
Now, you can install a Linux distribution on WSL. To do so, click on the Microsoft Store and type Ubuntu in its search box. Microsoft Store has different versions of Ubuntu. Choose the latest LTS version of Ubuntu and click “get”. Windows will automatically installs Ubuntu on the Linux subsystem.
After downloading from the Microsoft Store, you can open it from the Start menu. However, before launching it, update the Windows Subsystem for Linux (WSL). To do this, open Windows PowerShell or Command Prompt and type `wsl.exe –update`. It’s essential to update WSL, as it may not function properly without the latest updates.

Open Ubuntu from the start menu. If you receive the CPU virtualisation error, the CPU virtualisation of your system is disabled in your motherboard settings, and you must enable it. You can find many online guides on how to enable virtualization in the system BIOS.
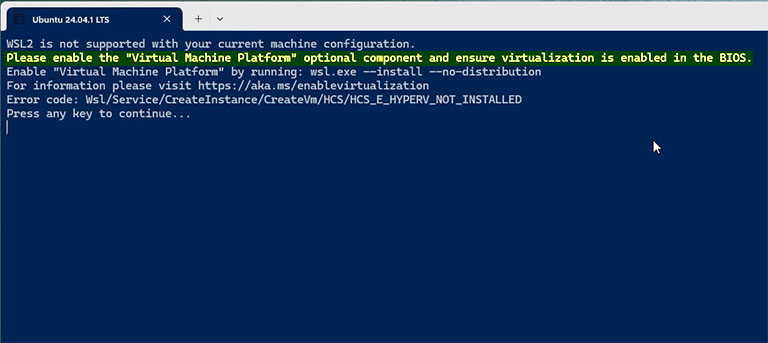
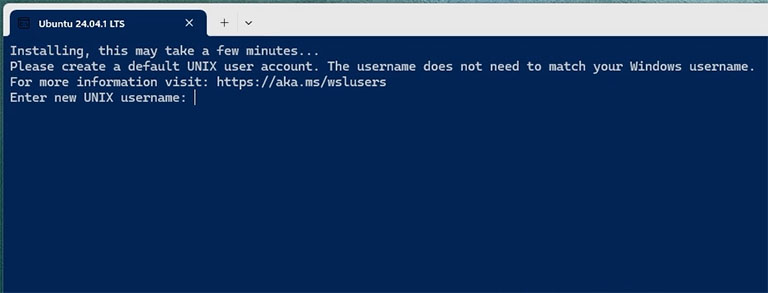
After enabling the CPU virtualisation, open Ubuntu from the Start menu again. It takes a few minutes to configure Ubuntu the first time you launch it, and then you will be asked to set up your username and password. Then, you can enjoy a brand new Linux inside your Windows system.
Install GROMACS on Windows Subsystem for Linux using Ubuntu package manager
A package manager is a program that automatically downloads and installs software from online repositories, making installation straightforward. The package manager for Ubuntu is called the Advanced Package Tool (APT).
To start, update Ubuntu’s package index list by executing the following command in WSL.
sudo apt update
After updating the package manager database, you can install GROMACS using the following command.
sudo apt install -y gromacs
To verify that GROMACS is installed on your system and to locate the GROMACS binary, execute the following command:
whereis gmx
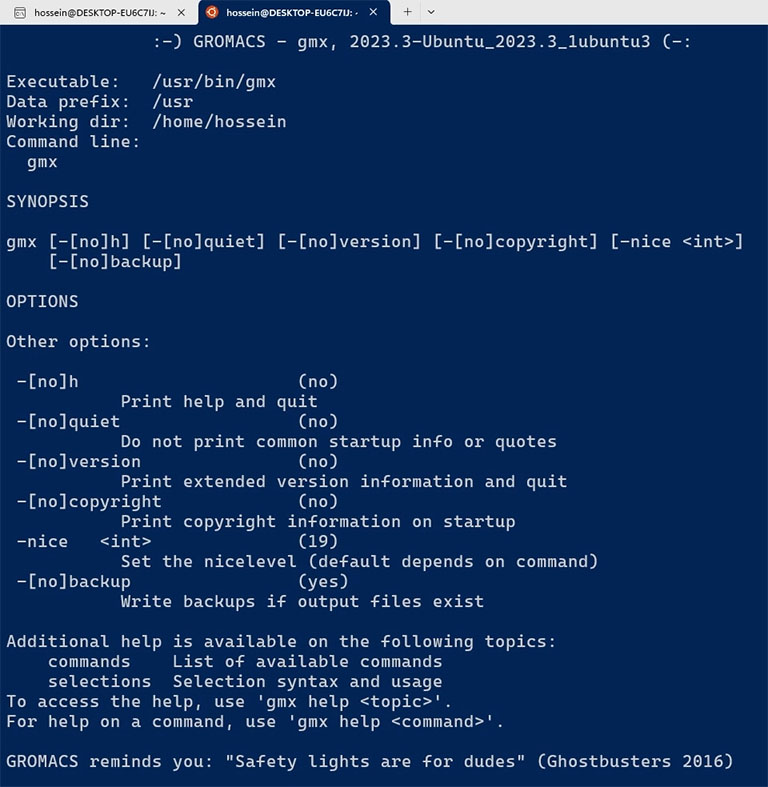
You can also check the GROMACS version by running the ‘gmx’ command.
gmx
Compiling GROMACS on Windows Subsystem for Linux
Compiling GROMACS’s source code is another method of installing it. It is arguably the most challenging method. So why would someone bother applying this method? There are three possible answers to this question.
- You may want to compile GROMACS with special features, like the ability to run on GPUs.
- You want to have the latest version of GROMACS.
- You may have a particular Hardware or operating system with no precompiled binary file for them.
To compile GROMACS, you need to obtain its source code. GROMACS is free and open-source software, which can be downloaded from its official website.
Copy the link to the latest GROMACS source code and download it using the wget command.
wget -c https://ftp.gromacs.org/gromacs/gromacs-2024.4.tar.gz
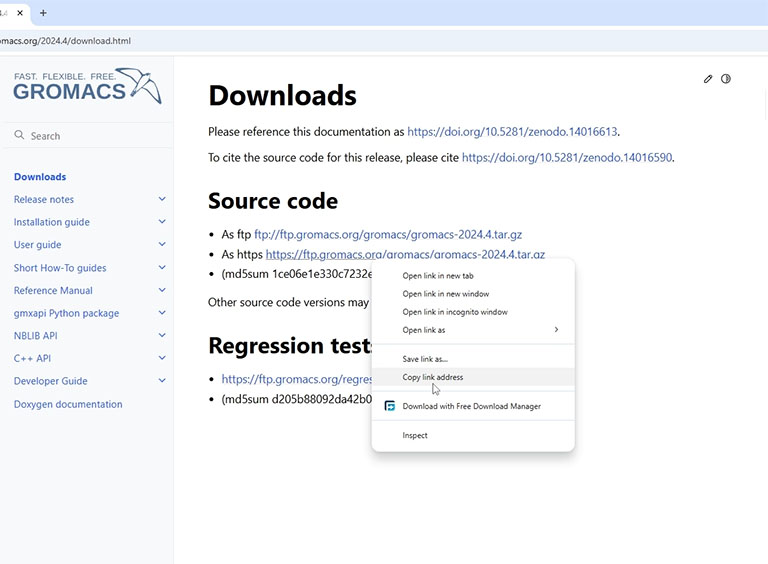
The GROMACS source code is provided in a compressed file. To extract it, use the tar command.
tar -xvzf gromacs-2024.4.tar.gz
Before compiling GROMACS, you need to install a compiler and other necessary tools. You can easily install them using the following command.
sudo apt install -y build-essential gcc vim cmake libpomp-dev hwloc libhwloc-dev
Now, create a new folder inside the GROMACS directory using the mkdir command and navigate to the build folder using the cd command.
mkdir gromacs-2024.4/build
cd gromacs-2024.4/build
To compile the code, first, you must configure the source code using the ‘cmake’ command. Pay attention to the ‘DCMAKE_INSTALL_PREFIX=/opt/gromacs-24.4’ option. This option specifies the location of the GROMACS binary file after compilation. If you have downloaded a different version of GROMACS, change the version number in the command.
cmake .. -DGMX_BUILD_OWN_FFTW=ON -DCMAKE_INSTALL_PREFIX=/opt/gromacs-24.4 -DCMAKE_C_COMPILER=gcc -DGMX_HWLOC=ON
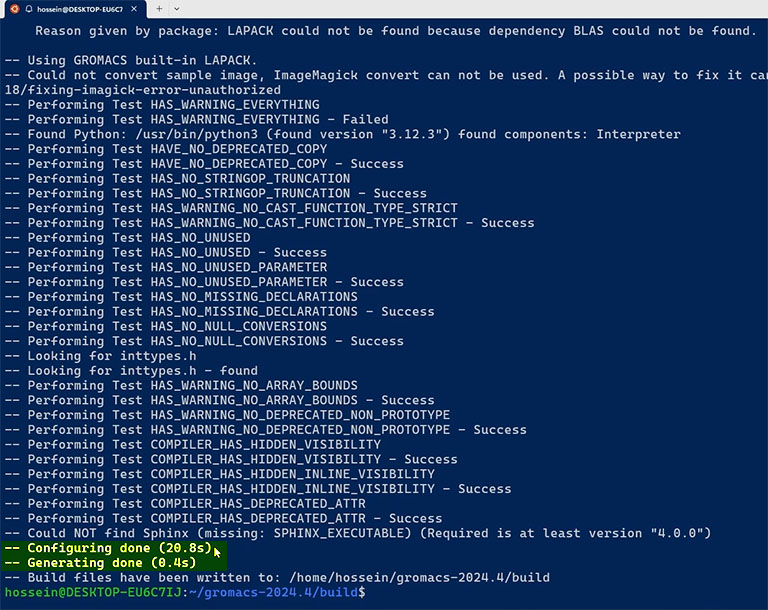
If the configuration was successful and you didn’t get any errors, you can compile the code using the make command. Put the number of desired CPU cores in the following command after the -j option.
make -j 4
If the compilation was successful without any error, use the following command to install the compiled binary files in the specified folder.
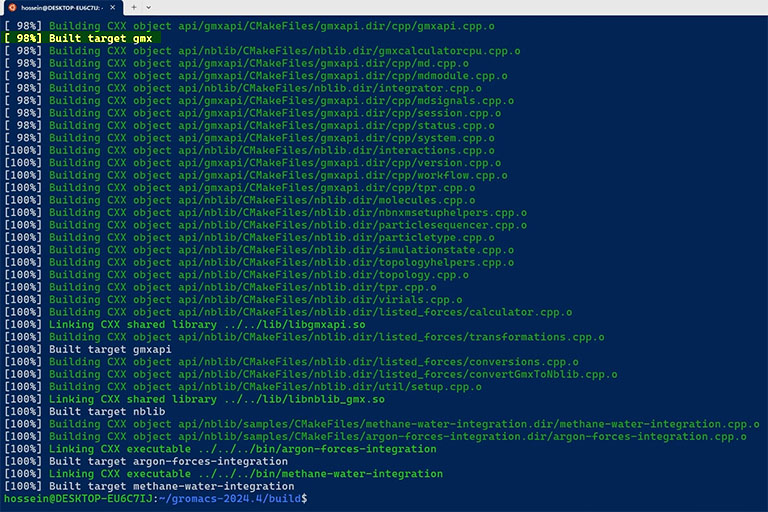
sudo make install
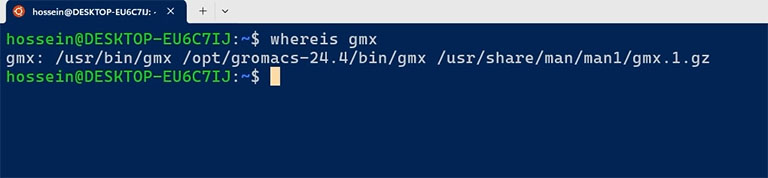
After installation, ensure the GROMACS binary files are in the OPT folder using the ‘where’ command.
whereis gmx
To access the GROMACS command in any directory, we need to source the file GMXRC.bash in the ‘.bashrc’ file. To do so, use the following command.
Change the version number and the path of the file if needed.
echo 'source /opt/gromacs-24.4/bin/GMXRC.bash' >> ~/.bashrc
Restart the terminal and execute the ‘gmx’ command. If you have done everything correctly, you should get the following output.
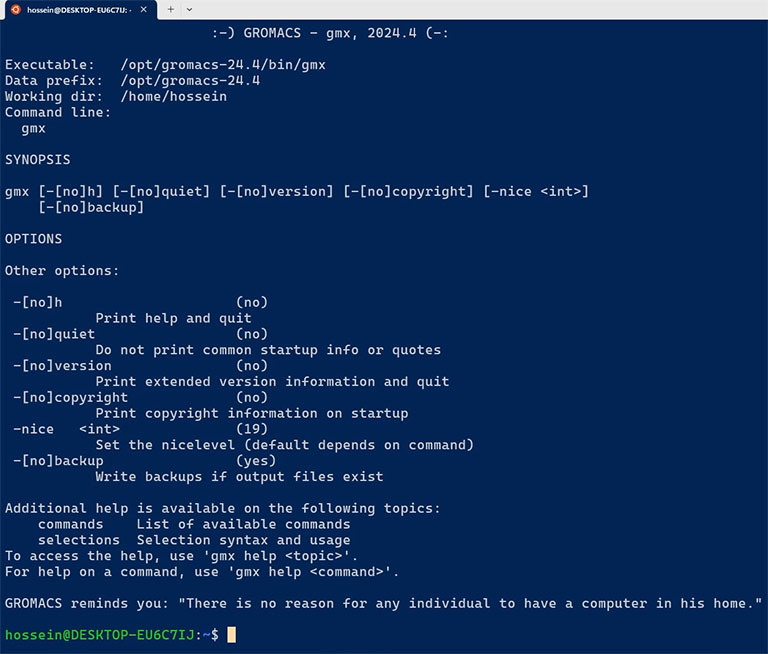
A note on compiling GROMACS with GPU support for Windows Subsystem for Linux
You can compile a version of GROMACS that runs simulations on graphic cards in Windows Subsystem for Linux. All you need to do is install the CUDA toolkit and compile GROMACS using the ‘GPU=CUDA’ option. However, based on my testing, I do not recommend this because the performance of GPU simulations on WSL is poor. Using GPUs in WSL can result in longer simulation times.
If you have a CUDA-enabled graphics card and are interested in running simulations on GPUs, I have another tutorial that shows how to create a GPU-enabled version of GROMACS on a Linux system.
installing VMD and accessing WSL files
VMD is a graphical program used to visualise molecular systems, and we can use it as a complement to GROMACS. We can not install VMD on WSL because it has no GUI; WSL only provides a text-based interface. However, We can install VMD directly on the host Windows instead of WSL.
VMD is free and open-source software that you can download from its official website. After downloading, install it like any other typical Windows program.
The installation is easy. However, a common cause of confusion is accessing WSL files from Windows to use programs like VMD. Actually, you cannot access WSL files directly from Windows. Instead, you can access Windows files and folders from within WSL. To access the Windows file system, you must go to ‘/mnt’, and then you can find Windows drivers.
For example, if you create a folder named WSL in the c driver of your system, you can access it using the following command in WSL.
cd /mnt/c/WSL
So, the best practice is to create a specific folder within one of your system drives and perform all simulations inside it. This approach makes it easy to access simulation files in Windows.
Running a test GROMACS simulation on Windows Subsystem for Linux
To run GROMACS simulations, you need several input files. I have created a sample simulation for you to test the installation on your system.
Download the sample input file from here and put it in the WSL folder you have created in the previous step.
This is a compressed file. So first, you need to uncompress it using the ‘tar’ command. Execute the following command in the WSL folder. And then go inside the uncompressed folder.
tar -xvzf gromacs_running_test.tgz
cd gromacs_running_test/
To run a simulation with GROMACS, you first need to create a specific binary input file in TPR format. Execute the following grompp command to do so.
gmx grompp -f nvt.mdp -p topol.top -c em.gro -o nvt.tpr -r em.gro -maxwarn 1
The ‘grompp’ command produces a file named ‘nvt.tpr’ file.
Finally, if everything is fine, you can run a GROMACS simulation using the ‘mdrun’ command.
gmx mdrun -v -deffnm nvt
Follow me on LinkedIn
Downloads
GROMACS is a free software you can download its source code from the GROMACS website.
To get the GROMACS test simulation input files, click the button below.
the file on your link is not working/corrupt. please fix it.
You have to download it directly in *.tgz file.
Not in zip file