Online GROMACS Workshop

GROMACS is a popular open-source molecular dynamics simulation software that can simulate the behaviour of complex molecular systems. GROMACS is well-known for its high performance and efficiency, making it an ideal tool for simulating various chemical and biochemical systems.
This workshop will teach you how to conduct molecular dynamics simulations on various systems and perform insightful analyses.
This workshop is suitable for those unfamiliar with molecular dynamics and the GROMACS software. All calculations will be taught from the beginning, along with the necessary theoretical background. However, experienced GROMACS users will also find many valuable topics in this workshop.
How the workshop will be conducted
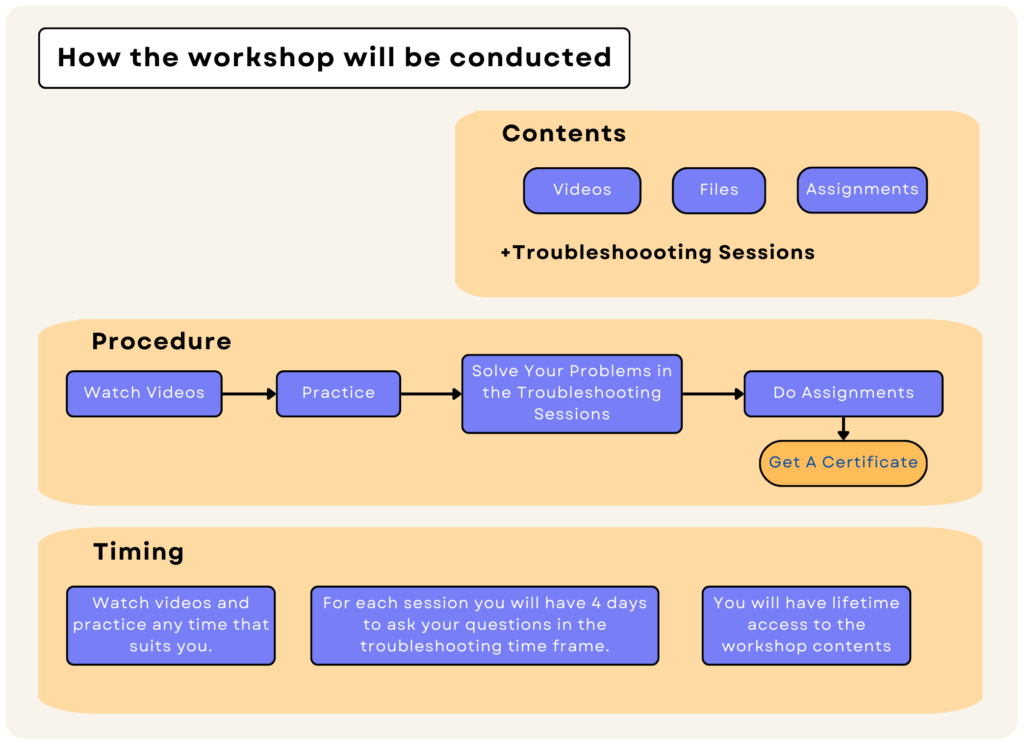
Molecular Dynamics is a complicated art that needs training and practice, as well as a solid theoretical foundation. Also, you may encounter many problems in practice. We know this as a fact. Thus, the inSilicoSci GROMACS workshop is more than just an online course.
Our GROMACS workshop has two major components: online content and troubleshooting sessions. The online content includes video tutorials and some assignments. In each session, we first explain the theory behind the computational methods, and then we use those methods in practice. At the end, there are a few exercises to solidify your understanding. You can expect an average of one hour of video in each session. In addition, each session includes a four-day troubleshooting meeting during which you can discuss any problems that may have occurred during that session. Each session also has assignments to solidify your learning. By submitting %65 of them correctly, you will receive a certification.
Schedule
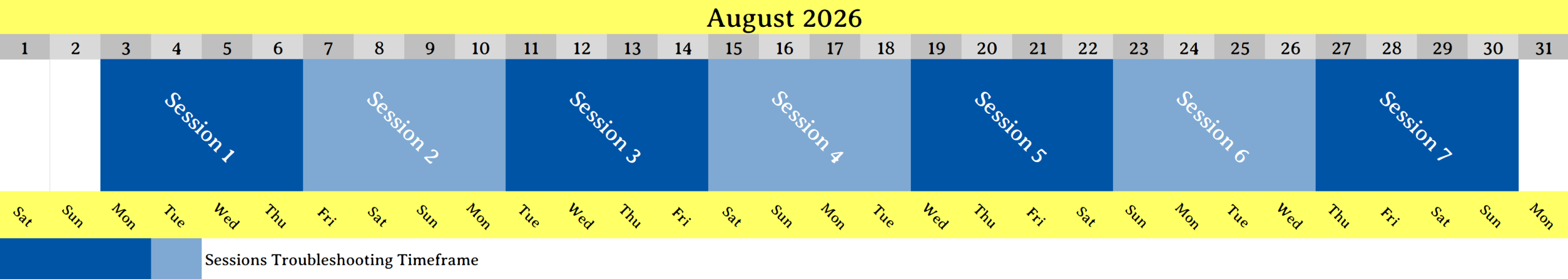
This GROMACS workshop is scheduled to take place from August 3 to 30, 2026.
Syllabus
Session One: Getting Started with Molecular Dynamics Simulation
- What is Molecular Dynamics Simulation?
- Introduction to Molecular Dynamics Force Fields
- Introduction to Molecular Dynamics Integration Schemes
- Introduction to Molecular Dynamics Ensembles
- Introduction to Periodic Boundary Conditions
- Installing GROMACS and VMD on Linux
- Installing GROMACS and VMD on Windows
Session Two: A Simple Protein MD Simulation
- How to Find and Download Protein Structures
- Introduction to the PDB File Format
- Visualising a Protein Structure Using VMD
- Removing Water Molecules and Fixing Missing Atoms in PDB Files
- Overview of the GROMACS Input Workflow
- Creating a Topology File for a Protein Structure
- Understanding GROMACS Topology (.top) and Coordinate Files
- Setting Up a Periodic Boundary Condition (PBC) Box
- Adding Solvent Molecules and Ions to the Simulation Box
- Energy Minimisation of the Molecular System
- Equilibrating the System in the NVT Ensemble
- Introduction to Position Restraints in GROMACS
- Understanding the GROMACS MDP (Molecular Dynamics Parameters) File
- How to Check If the System Has Reached Equilibrium
- Equilibrating the System in the NPT Ensemble
- Running the Production Simulation
- Understanding GROMACS Output Files
- Restarting and Extending a Simulation
- Visualising and Inspecting Trajectory Files Using VMD
- Performing Basic Analysis of Simulation Results
- Performing and Interpreting RMSD (Root Mean Square Deviation) Analysis
- Performing and Interpreting Radius of Gyration Analysis
Session Three: A Protein-Ligand Simulation
- Common Challenges in Protein–Ligand Complex Simulations
- Installing and Introducing the pdbfixer Tool
- Introduction to the CGenFF Force Field Generator
- Preparing a Ligand Structure for CGenFF
- Interpreting CGenFF Penalty Scores
- Generating GROMACS Topology Files from CGenFF
- Running a Test Simulation to Validate Ligand Topology
- Generating Protein Topology Using the CHARMM36 Force Field
- Combining Protein and Ligand Topology Files
- Converting GROMACS Topology Files to .itp Format
- Troubleshooting Common Errors in Protein–Ligand Simulations
- Creating a New Index Group Using gmx make_ndx
- Generating Position Restraints for the Ligand
- Analysing Protein–Ligand System Stability Using RMSD
- Introduction to Principal Component Analysis (PCA)
- Performing PCA on a Protein–Ligand Complex
- Plotting and Interpreting the Free Energy Landscape
Session Four: Dealing with Metallic Ligands and Molecules
- Introduction to AmberTools and the MCPB.py Workflow
- Installing AmberTools24 via Miniconda
- Preparing a Protein Structure for Use with MCPB.py
- Rebuilding a Ligand Structure for MCPB.py Compatibility
- Preparing a Metal Ion for MCPB.py Processing
- Performing DFT Calculations to Derive Missing Parameters
- Generating Topology and Coordinate Files Using the tLeap Program
- Converting AMBER Topology and Coordinate Files to GROMACS Format Using the Acpype Tool
- Running a Test Simulation in GROMACS to Validate Setup
Session Five: Bio-Membrane Simulations
- Introduction to the CHARMM-GUI Web Application
- Building a Simple Lipid Membrane System
- Conducting a Membrane System Simulation Using GROMACS
- Constructing an Asymmetrical Bilayer Containing Glycolipids
- Building a Membrane with a GPI-Anchored Protein
- Constructing a Membrane Containing a G-Protein-Coupled Receptor (GPCR)
Session Six: Calculating Free Energy
- Introduction to Alchemical Methods for Calculating Free Energy
- Overview of Geometrical Methods for Free Energy Calculation
- Explaining End-State Methods for Free Energy Calculation
- Introduction to the MM-PBSA Method for Free Energy Estimation
- Installing the gmx_mmpbsa Program
- Calculating Protein–Ligand Binding Energy Using the MM-PBSA Method
- Introduction to Free Energy Perturbation (FEP) Methods
- Calculating Protein–Ligand Binding Energy Using the FEP Method
Session Seven: Umbrella Sampling and Free Energy Calculations in GROMACS
- Introduction to the pull code and the Umbrella Sampling (US) method
- Explanation of the Weighted Histogram Analysis Method (WHAM)
- Preparing the MDM2–p53 complex for US simulations
- Reorienting the complex along the z-axis to define the reaction coordinate
- Running a pulling simulation to generate the reaction coordinate
- Extracting representative frames from the pulling trajectory
- Setting up and running multiple Umbrella Sampling simulations
- Performing WHAM analysis and combining simulation windows
- Interpreting WHAM histograms and assessing sampling quality
- Adding extra windows to improve convergence in poorly sampled regions
- Plotting and interpreting the Potential of Mean Force (PMF) diagram
- Calculating free energy differences from the PMF
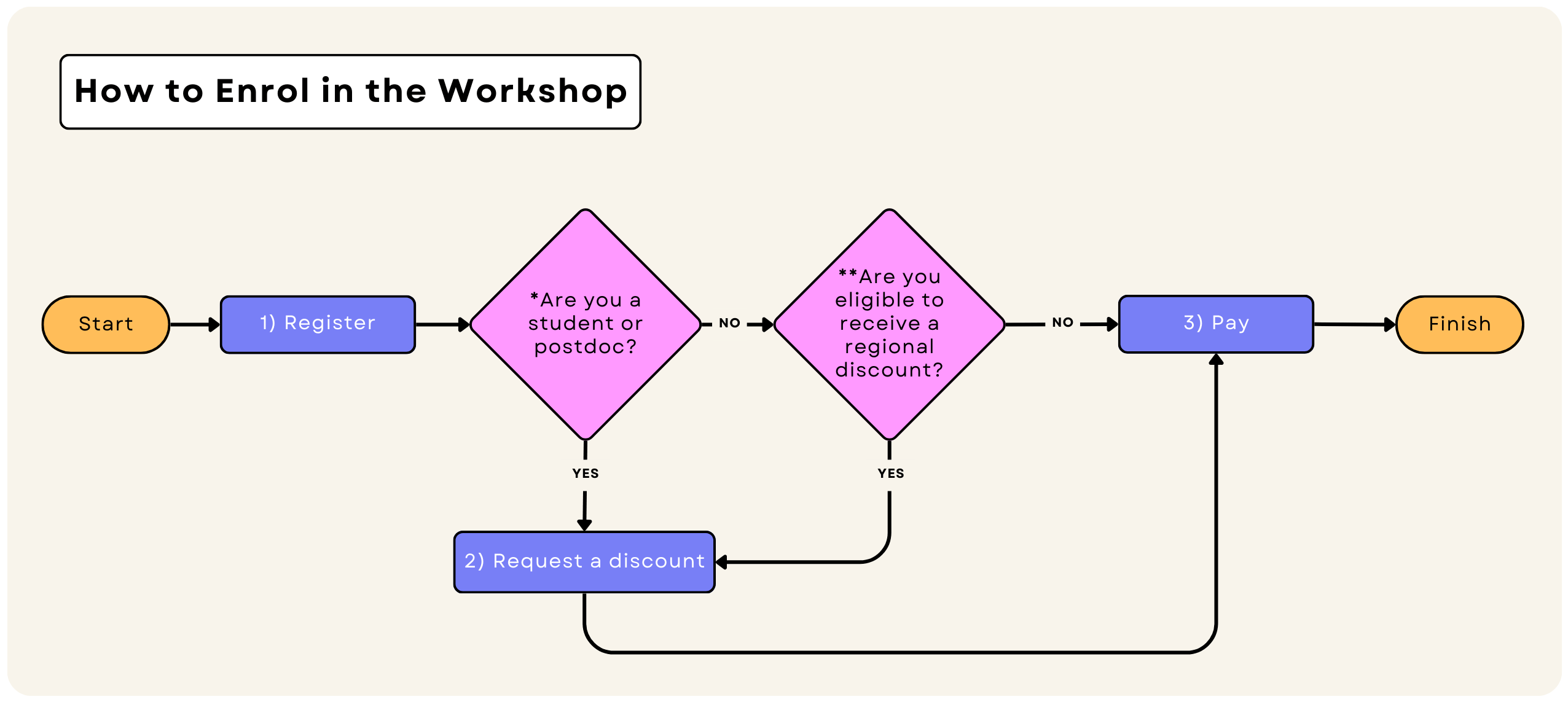
How to enrol in the workshop
Pricing
Students and Postdocs
Others
129.00 USD
199.00 USD
- To use the student & postdoc plan, You need to provide us with a valid student ID card or any document that proves your student or postdoctoral status.
- Also, depending on your location, you may qualify for regional discounts. Check the regional discounts section to see how much you can save.
Regional Discounts
We are not communists, but we believe everyone should have the opportunity to learn and thrive, so we offer discounts based on purchasing power in various countries to make education accessible.
- To receive a discount for a specific country, you need to be a resident of that country.
- If you enrolled with a discount code, do not use a foreign VPS/VPN while participating in the workshops.
- To receive a discount code, please send us an image of your student card or any document showing you are currently studying or residing in a specific country.
- There is no need for an explanation. Just send your document via WhatsApp or email and introduce yourself.
- Please refer to the list below to check how much discount you can receive.
- Afghanistan %60
- Albania %40
- Algeria %50
- Andorra %0.0
- Angola %30
- Argentina %60
- Armenia %40
- Australia %0.0
- Austria %0.0
- Azerbaijan %30
- Bahamas %0.0
- Bahrain %0.0
- Bangladesh %50
- Barbados %0.0
- Belarus %50
- Belgium %0.0
- Belize %0.0
- Benin %50
- Bhutan %60
- Bolivia %40
- Bosnia and Herzegovina %40
- Botswana %30
- Brazil %20
- Brunei %0.0
- Bulgaria %30
- Burkina Faso %50
- Burundi %60
- Cabo Verde %30
- Cambodia %50
- Cameroon %40
- Canada %0.0
- Central African Republic %30
- Chad %30
- Chile %20
- China (Mainland) %20
- Colombia %50
- Comoros %40
- Congo (Brazzaville) %0
- Congo (Kinshasa) %30
- Costa Rica %20
- Croatia %30
- Cuba %60
- Cyprus %0.0
- Czechia %0.0
- Denmark %0.0
- Djibouti %20
- Dominica %0.0
- Dominican Republic %30
- East Timor %30
- Ecuador %20
- Egypt %60
- El Salvador %30
- Equatorial Guinea %30
- Eritrea %60
- Estonia %0.0
- Eswatini %40
- Ethiopia %50
- Fiji %40
- Finland %0.0
- France %0.0
- Gabon %20
- Gambia %50
- Georgia %50
- Germany %0.0
- Ghana %50
- Greece %0.0
- Grenada %0.0
- Guatemala %20
- Guinea %30
- Guinea-Bissau %50
- Guyana %30
- Haiti %20
- Honduras %30
- Hungary %30
- Iceland %0.0
- India %60
- Indonesia %50
- Iran %60
- Iraq %20.0
- Ireland %0.0
- Israel %0.0
- Italy %0.0
- Ivory Coast %40
- Jamaica %20
- Japan %0.0
- Jordan %40
- Kazakhstan %40
- Kenya %40
- Kiribati %0.0
- Kosovo %40
- Kuwait %0.0
- Kyrgyzstan %60
- Laos %60
- Latvia %0.0
- Lebanon %60
- Lesotho %40
- Liberia %40
- Libya %60
- Liechtenstein %0.0
- Lithuania %0.0
- Luxembourg %0.0
- North Macedonia %50
- Madagascar %60
- Malawi %40
- Malaysia %40
- Maldives %30
- Mali %50
- Malta %0.0
- Marshall Islands %0.0
- Mauritania %50
- Mauritius %40
- Mexico %20
- Micronesia %0.0
- Moldova %40
- Monaco %0.0
- Mongolia %50
- Montenegro %40
- Morocco %40
- Mozambique %40
- Myanmar %60
- Namibia %30
- Nauru %0.0
- Nepal %60
- Netherlands %0.0
- New Zealand %0.0
- Nicaragua %50
- Niger %50
- Nigeria %40
- North Korea
- Norway %0.0
- Oman %0.0
- Pakistan %60
- Palau %10
- Panama %0.0
- Papua New Guinea %0.0
- Paraguay %40
- Peru %30
- Philippines %50
- Poland %30
- Portugal %20
- Qatar %0.0
- Romania %40
- Russia %30
- Rwanda %50
- Saint Kitts and Nevis %0.0
- Saint Lucia %0.0
- Saint Vincent and the Grenadines %20
- Samoa %0.0
- San Marino %0.0
- Sao Tome and Principe %0.0
- Saudi Arabia %0.0
- Senegal %40
- Serbia %40
- Seychelles %30
- Sierra Leone %60
- Singapore %0.0
- Slovakia %0.0
- Slovenia %0.0
- Solomon Islands %0.0
- Somalia %50
- South Africa %30
- South Korea %0.0
- South Sudan
- Spain %0.0
- Sri Lanka %60
- Sudan %60
- Suriname %50
- Sweden %0.0
- Switzerland %0.0
- Syria %60
- Taiwan %0.0
- Tajikistan %60
- Tanzania %40
- Thailand %50
- Togo %50
- Tonga %0.0
- Trinidad and Tobago %0.0
- Tunisia %60
- Turkey %40
- Turkmenistan
- Tuvalu %0.0
- Uganda %40
- Ukraine %40
- United Arab Emirates %0.0
- United Kingdom %0.0
- United States %0.0
- Uruguay %0.0
- Uzbekistan %60
- Vanuatu %0.0
- Venezuela %60
- Vietnam %50
- Yemen %60
- Zambia %40
- Zimbabwe %20
Sample Video
Watch the sample video on YouTube.
https://youtu.be/-S1eP-iWE8c
Certification
After finishing the GROMACS workshop, you can apply for certification. The certification does not include a grade and only confirms that you participated in the GROMACS workshop. The certification requirement is that you complete all the sessions and submit at least 80 percent of the assignments correctly.