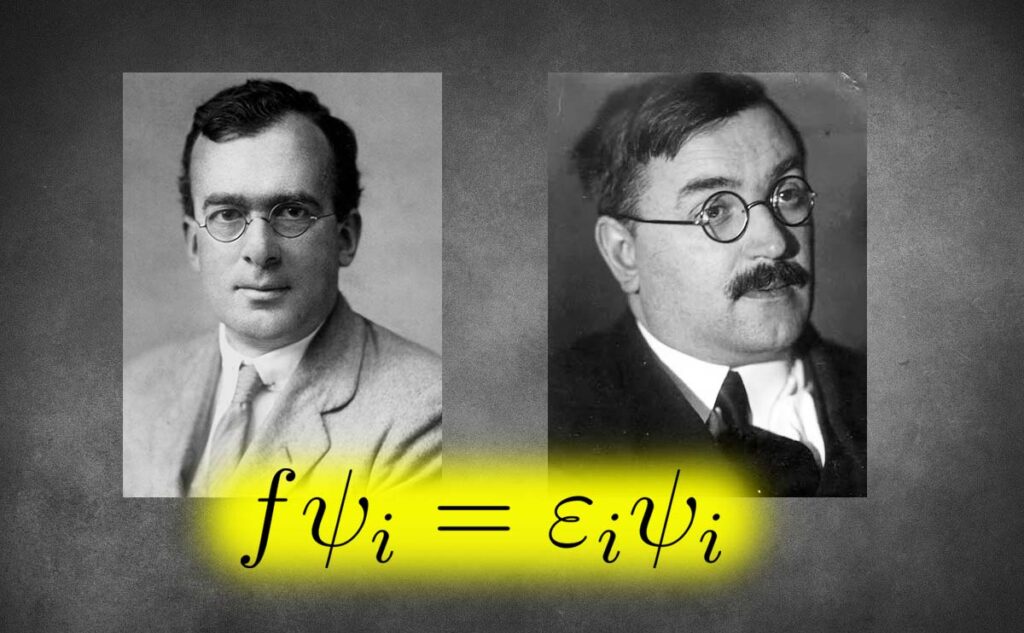



















Generally, having N electrons and M spin-orbitals, we can construct ![]() different Slater determinants. However, the Hartree-Fock approach only uses one of them called the Hartree-Fock ground state determinant. The other determinants can represent the excited states of the electronic system. Also, a linear combination of these determinants is a more accurate wave function, which more precisely considers the correlation between electrons. A more elaborate and computationally cumbersome method, configuration interaction (CI), utilises the linear combination of these determinants to describe electronic systems. Using a single Slater determinant arguably equals treating electron-electron repulsion in an average way, which is the essence of the Hartree-Fock approximation.
different Slater determinants. However, the Hartree-Fock approach only uses one of them called the Hartree-Fock ground state determinant. The other determinants can represent the excited states of the electronic system. Also, a linear combination of these determinants is a more accurate wave function, which more precisely considers the correlation between electrons. A more elaborate and computationally cumbersome method, configuration interaction (CI), utilises the linear combination of these determinants to describe electronic systems. Using a single Slater determinant arguably equals treating electron-electron repulsion in an average way, which is the essence of the Hartree-Fock approximation.




































The coulomb operator has a straightforward interpretation. The exact Coulomb interaction between two electrons in the atomic unit is r12-1. Likewise, in the Hartree-Fock approximation, electron one in χa experiences a one-electron potential from χb equal to ![]() and because in equation 38, the summation is over all spin-orbitals, each electron feels Coulomb repulsion from all other electrons. Note that each integral in the summation is independent of the others, which is why the Hartree Fock approximation does not account for the Coulomb correlation between electrons.
and because in equation 38, the summation is over all spin-orbitals, each electron feels Coulomb repulsion from all other electrons. Note that each integral in the summation is independent of the others, which is why the Hartree Fock approximation does not account for the Coulomb correlation between electrons.
On the other hand, the exchange operator has no perceivable classical interpretation and results from exchange-correlation between electrons with parallel spins, which I previously explained. The existence of the exchange operator in the Hartree Fock equation implies that the Hartree Fock method is not entirely uncorrelated and considers the correlation between electrons with parallel spins. Using the Coulomb and exchange operators, we can shorten equation 38 as:











































Congratulation!
As far as I can tell, this is the simplest text for learning one of the most difficult concepts in computational chemistry.
Excellent explanation about the Hartree Fock method; thank you for shear.
Excellent!!!
Could you give a similar explanation for DFT ?
Thank you Zhang. Acctually I’m working on it.
Looks neat, but the equations and figures are not showing currently (tested firefox, chrome and mobile). Hopefully the fix is easy, I’m looking forward to reading this to help me with my computational physics course.
Thank you for informing us.
Would you please tell us from where you checked our website?
Is the equation not loading even in incognito mode?
Excellent explanations. So simply written and understandable. Keep doing this please
Keep up the good work!!! Wonderful
This is so helpful! I am doing a seminar on this and this is wonderful help!! This is the best explanation I have ever seen! Thank you.
This is truly one of the most complete ad easy to digest explanation on the HF method. Thank you, very helpful. If you could do the same for post HF methods like CASSCF it would be golden.
Excellent! Thank you.
This is brilliant; thank you!